
The genome contact map explorer: a tool to find patterns in piles of DNA contact data
The problem
Every chromosome in our cells is a long polymer that is packaged and folded in particular ways. Researchers don’t exactly know how, but it is clear that it is not a random folding process. Instead, it looks like chromosomes are folded into a network of 3D compartment of different sizes.
How and why our DNA is folded is crucial to understand, because the 3D folding patterns of DNA affect several of the cell’s biological processes, like how genes are regulated. For example, during embryo development, it is important that the correct sets of genes get turned on in the correct order and correct parts of the body. If this fails, the organism’s body plan might get screwed up. In many organisms, such as the fruit fly, this is solved with epigenetic marks – long stretches of specific chemical tags that cover the genes that are supposed to be off. And importantly, 3D folding patterns are often correlated (but not always) with the location of these marks. So, understanding the 3D organisation of DNA, could for example help us to better understand epigenetic mechanisms.
The solution
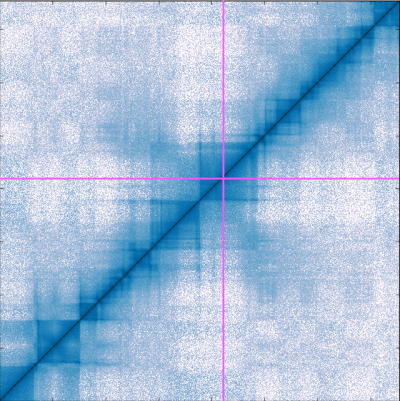
Building on Chromosome Conformation Capture experiments that detect physical encounters between two DNA fragments inside the cell, researchers created the Hi-C method – the first genome-wide method that gives a glimpse into DNA’s 3D structure. Hi-C finds the number of contacts between all DNA fragments in the nucleus (down to 1,000 base pairs), and the Hi-C data is often visualised as a heat map. However, as chromosomes are large, the number of contact pairs becomes huge, especially at high resolution. This causes practical problems when researchers want to see and interact with the map. To overcome this problem, we developed the Genome Contact Map Explorer, open-source software that gives researchers the ability to visually browse, scroll and zoom in on their Hi-C maps. Alongside the maps, it is also possible to add additional genetic data such as gene expression, protein binding, or epigenetic data. Apart from the browser, we also made a programmable interface that allow developers to design their own Hi-C analysis toolkit.
Browsing Hi-C maps in gcmapexplorer is fast because internally it never loads the complete Hi-C data into the computer’s memory. Instead, it only loads the parts that you see on the screen, and reloads new sections of the map when zooming and scrolling. To do this smoothly requires a suitable file format. Internally, the gcmapexplorer uses the hdf5 format which is much faster to read than a simple text file. Because of gcmapexplorer’s efficient use of memory, it is possible to load a large number of maps, for example from mutant and control experiments, and arrange them side by side.
The gcmapexplorer is open-source software written in python, and downloadable here. Currently gcmapexplorer is being used by molecular biologists in all parts of the world.
Read the original paper
Kumar, R., Sobhy, H., Stenberg, P., & Lizana, L. (2017). Genome contact map explorer: a platform for the comparison, interactive visualization and analysis of genome contact maps. Nucleic Acids Research, 45(17), e152.